Introducing the Super Deduper
The Super Deduper is a C/C++ application that was created to remove PCR duplicates from high throughput sequencing data. First I will give an overview on installation then go over how to run it and how it works. If you run into any problems or have any questions, please, submit an issue on the github page. I hope you find this application useful in your research.
Here is the basic installation and basic use case of the Super Deduper. It has been tested on Linux as well as OS X.
$ git clone git@github.com:dstreett/Super-Deduper.git
$ cd Super-Deduper
$ make
$ echo "Example of Paired End inputs (Two files [-1 and -2] and Interleaved [-i])"
$ ./super_deduper -1 ./examples/phix_example_PE1 -2 ./examples/phix_example_PE2
$ ./super_deduper -i ./examples/interleaved_example.fastq.gz
$ echo "Example of Single End Reads [-U]"
$ ./super_deduper -U ./examples/phix_example_SE.fastq.gz
How Super Deduper Works
The Super Deduper turns a region of the sequence (or the entire sequence) into a binary number. It does this by mapping A to 00, T to 11, C to 01, and G to 10.
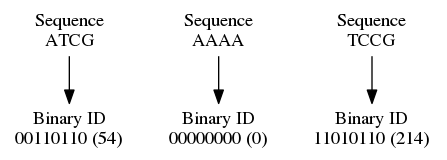
Super deduper concatenates read 1's and read 2's binary numbers together and creates a unique ID for the pair that is then used to determine duplications in a binary search tree. With single end reads the same thing happens, except there is no read 2 to concatenate with.
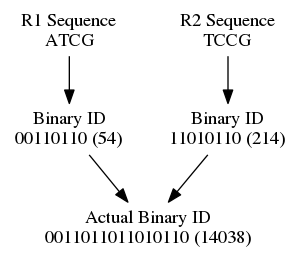
Both the read and the reverse compliment are tested for - however, only the larger of the two are kept. Doing this ensures that the reverse compliment is tested for and that all reads that are duplicates (be in a reverse compliment duplicate) are tested and mapped to the same unique ID. A node is created in this tree if a new sequence ID is seen. If an ID is all ready seen a summation of the quality score of the entire read is taken. The read with the higher summation is taken. The reason for a summation versus an average is summations give preferential treatment to longer reads as well as higher quality scores.
Arguements and what they do
Here are the basic arguements
If you are doing paired end reads (that aren't interleaved)
-1, --read1 [path to read 1]
-2, --read2 [path to read 2]
If you want to use an interleaved file - no problem
-i, --interleaved-input [path to interleaved file]Additionally, single ends are handled just fine
-U, --singleend [path to single end file]
All these options can be comma seperated to add multiple files. Additionally, fastq or fastq.gz are accepted as input.
To change the region in which the Super Deduper determines duplicates here are the arguments
-s, -start NUM [starting base pair of ID]
-l, --length NUM [number of base pairs from the starting base pair]
If you enter inputs that are out of bounds for reads the application will stop running and exit.
Output arguments are as follows
-p, --output-prefix PREFIX [This will add PREFIX to the start of "_nodup_PE1.fastq]
-o, --interleaved-output PREFIX [Outputs in interleaved format with prefix specified]
-g, --gzip-output [Outputs in gzipped format]
If you would like to save the duplication sceme for a later data, you can actually output the tree and input it the next time you run it.
-O, --output-tree PATH [outputs tree to the file]
-I, --input-tree PATH [Tree files to read in (comma seperated)]
Thank You
I would like to thank everyone who took the time to download and use this application. In efforts to continuely import and grow, let me know any issues you run into or if anything is unclear.